Die Tumore des zentralen Nervensystems werden entsprechend der weltweit gebräuchlichen und allgemein anerkannten WHO-Klassifikation der Hirntumore nach verschiedenen Kriterien eingeteilt und zugeordnet. Die Einteilung erfolgt erstens nach der mutmaßlichen Tumorzellenherkunft (benignes Muttergewebe), zweitens nach dem Grad der Bösartigkeit und letztens auch nach molekularen Eigenschaften (Gene, Gen-Amplifikation, -Deletion, -Translokation etc.).
Besonders relevant sind die feingeweblichen (pathohistologischen) Merkmale der Tumore, die an Schnittpräparaten des Tumorgewebes unter dem Mikroskop bestimmt werden. Hierzu werden Tumorgewebsproben entweder im Rahmen einer offenen Operation oder durch eine stereotaktische Biopsie gewonnen und analysiert.
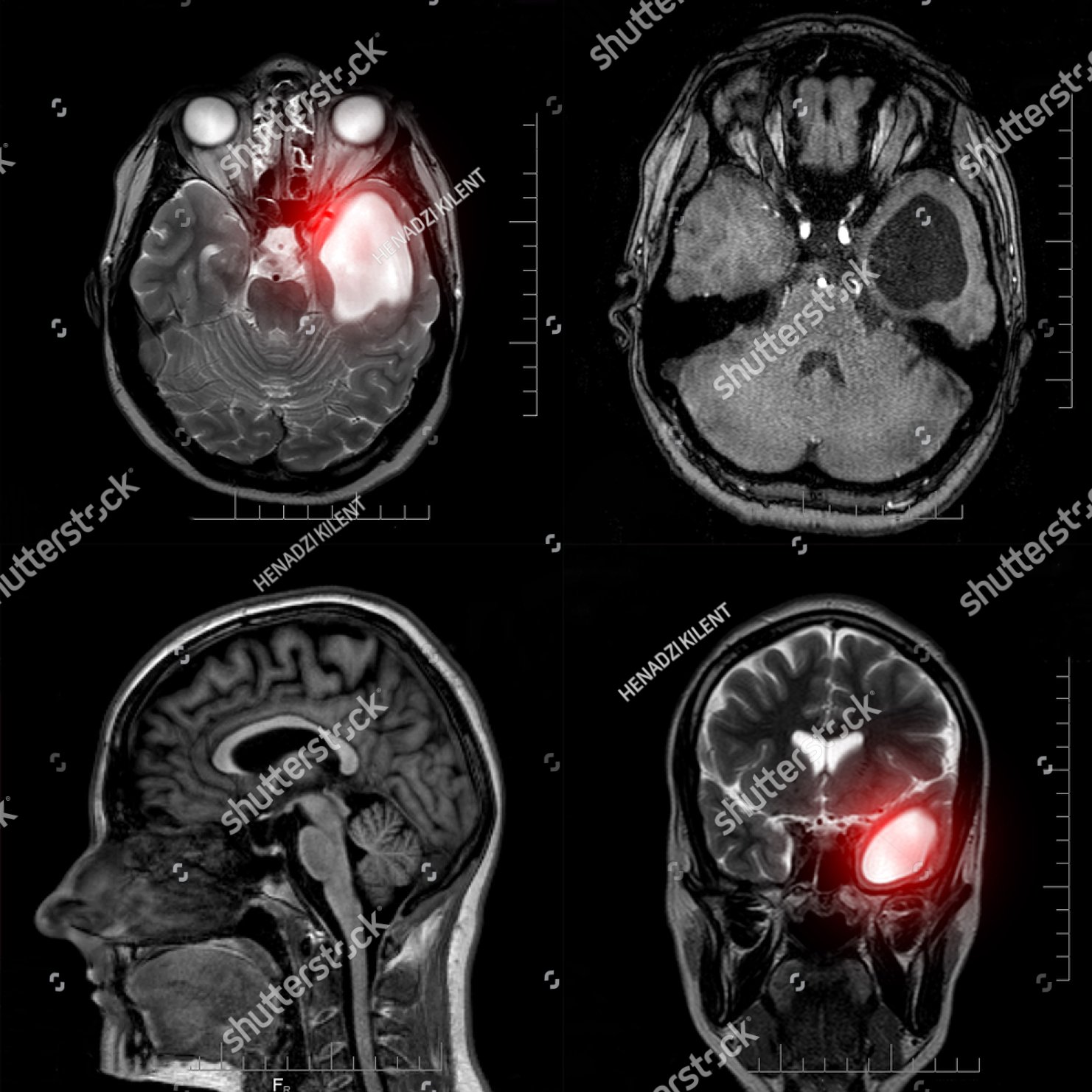
Zur Abklärung von Raumforderungen im Schädelinnenraum eignen sich die Computertomographie (CT) und in erster Linie die Magnetresonanztomographie (MRT).
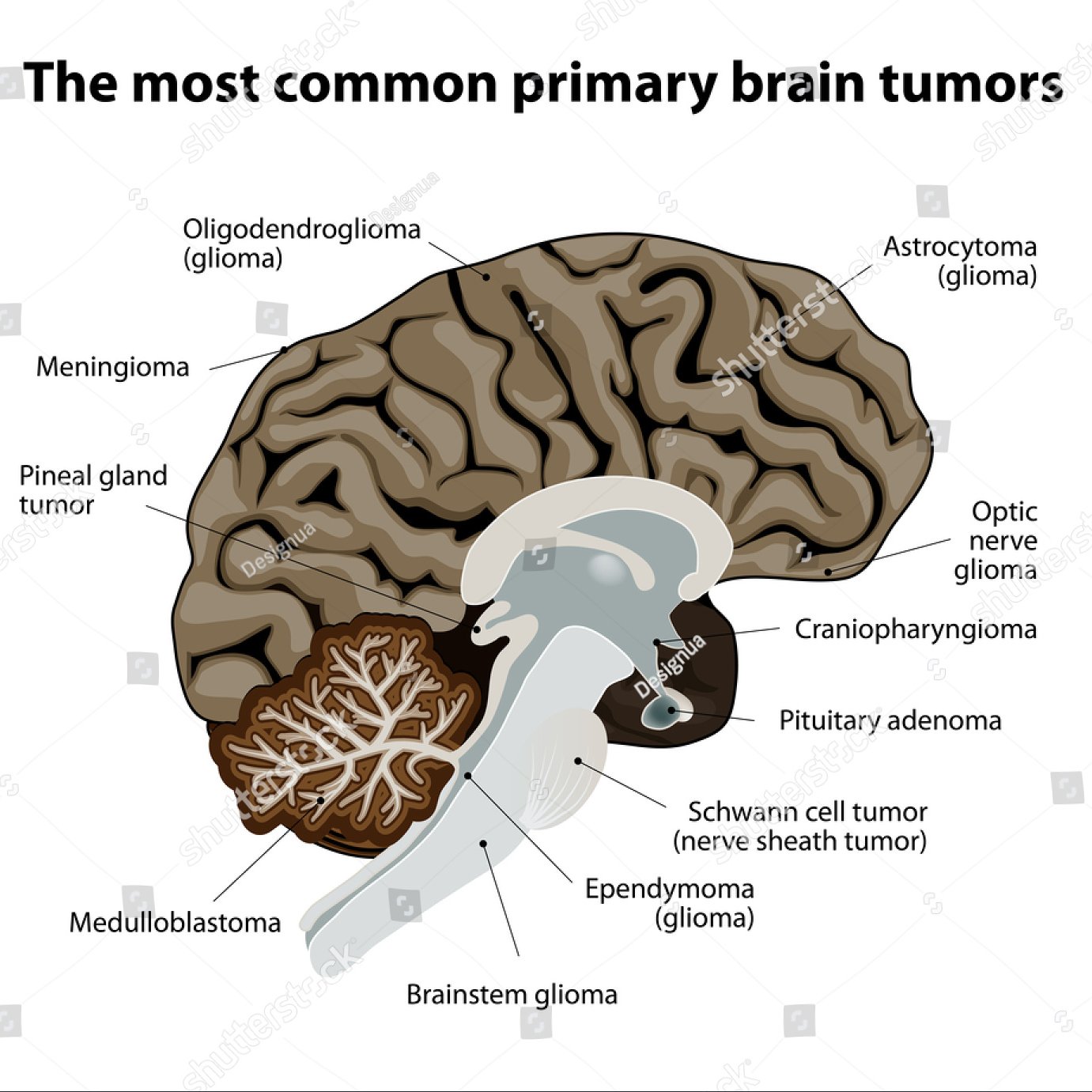
Gliome umfassen: Astrozytome, Glioblastome, Oligodendrogliome, Ependymome
KNOSP-KLASSIFIKATION
(Prof. Dr. Engelbert Knosp – 2002–2019 Vorstand der Wiener Universitätsklinik für Neurochirurgie)
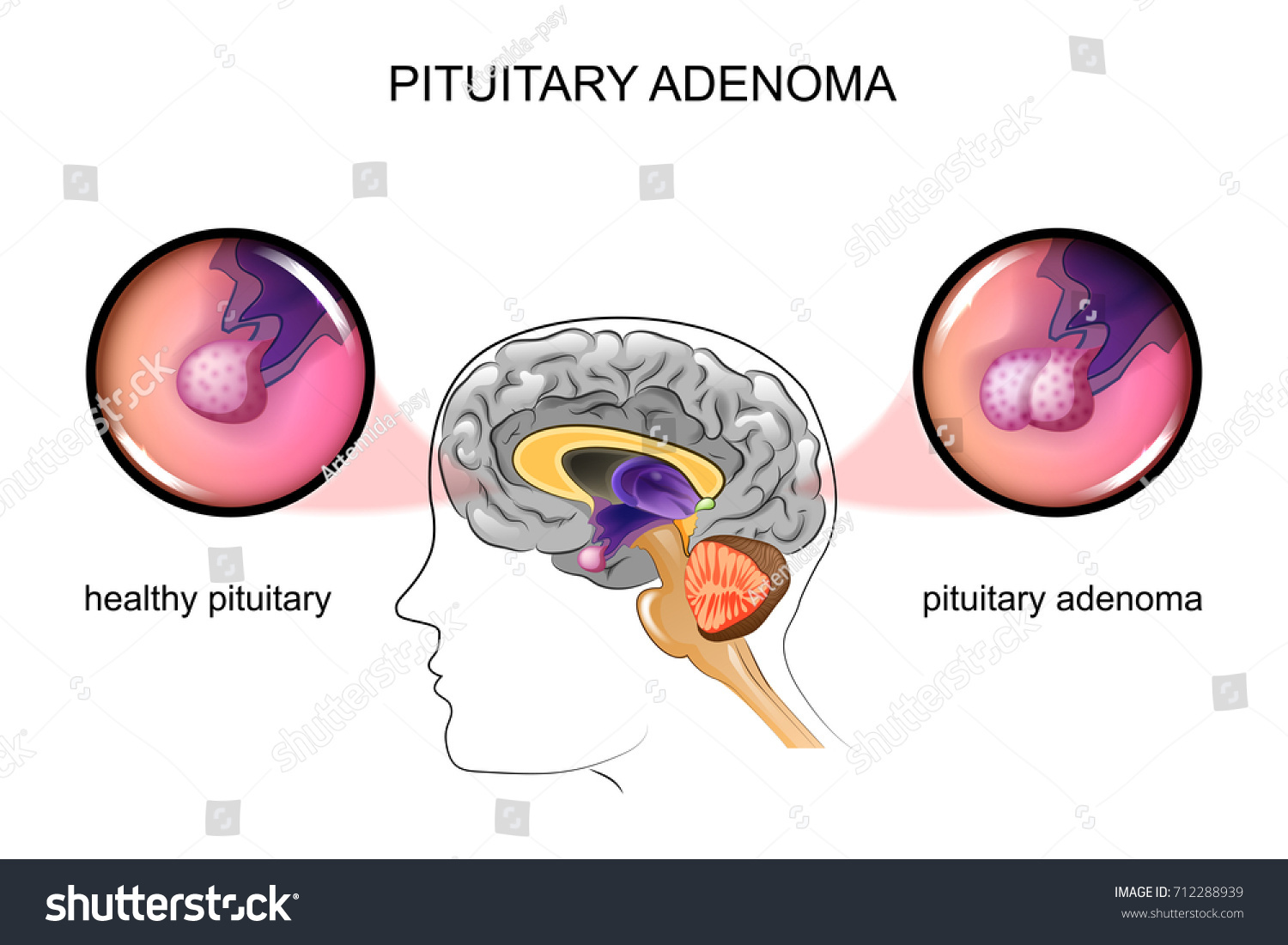
Seit 1993 wird die Knosp-Klassifikation der parasellären Ausdehnung von Hypophysenadenomen, basierend auf OP-Mikroskop-Daten, zur Einschätzung von Invasivität und Prognose international verwendet. Durch die endoskopische Operationstechnik ist nun intraoperativ eine direkte Sicht auf die mediale Wand des Sinus cavernosus möglich. Die Klassifikation wurde daher 2014 mit endoskopischen Daten ausgearbeitet und im Journal of Neurosurgery (Micko et al., 2015) publiziert.
Die Indikation zur Operation eines Hypophysenadenoms wird üblicherweise beim Vorliegen eines Chiasmasyndroms (Druck eines großen Tumors auf die darüberliegende Sehnervenkreuzung) oder eines übermäßigen Hormonexzesses gestellt, bzw. wenn es bereits zu einer Hypophysenvorderlappen-Insuffizienz (durch einen sogenannten Druck eines großen Tumors auf den gesunden Hypophysenteil) gekommen ist.
Prolaktinome werden als einzige Adenome primär medikamentös behandelt.
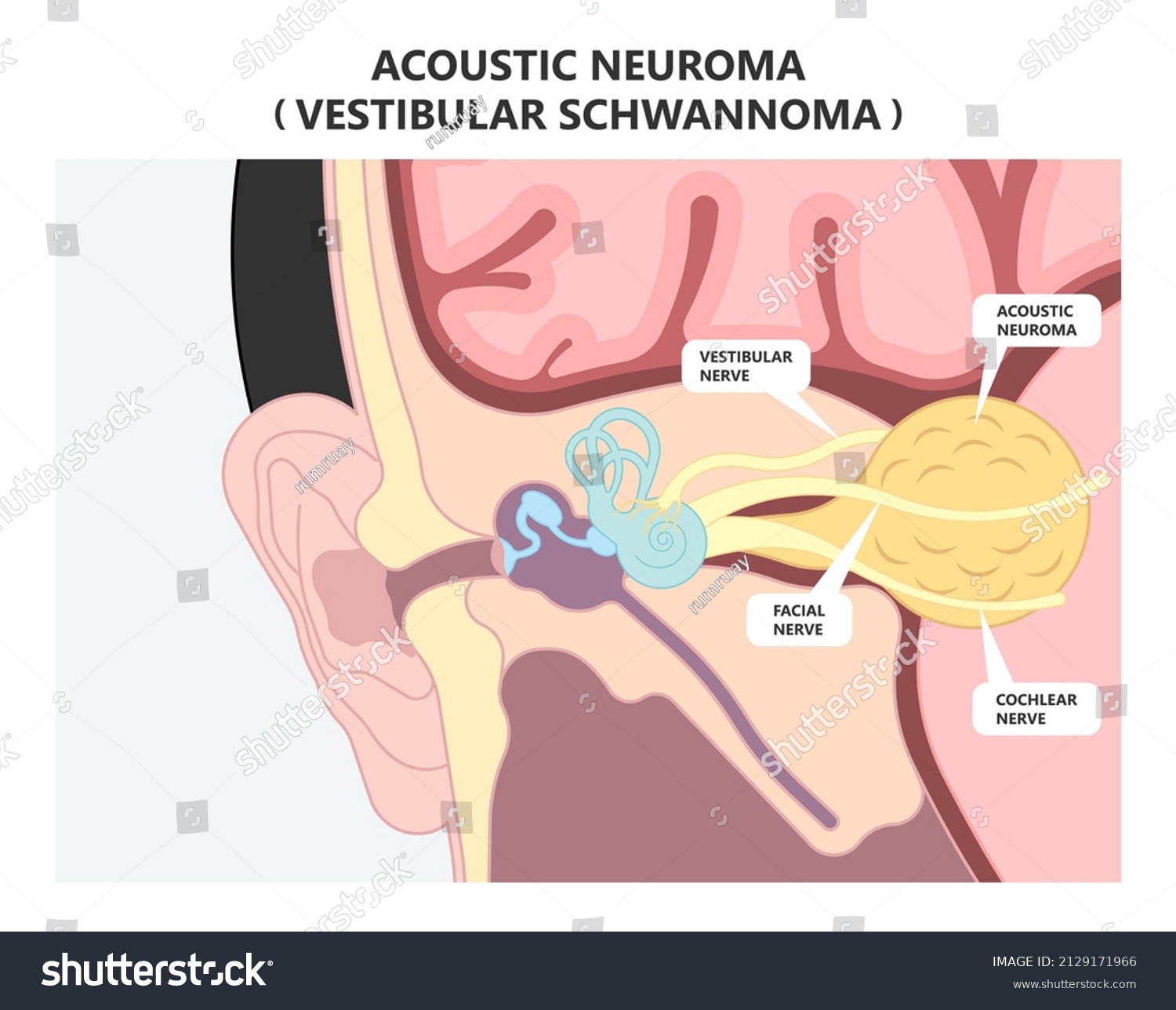
Hirntumorchirurgie
Chirurgischer Eingriff zur Entfernung eines Akustikneurinoms/Vestibularisschwannoms
(Operierender Neurochirurg: Univ.-Prof. Karl Rössler; Universitätsklinik für Neurochirurgie, 2021)
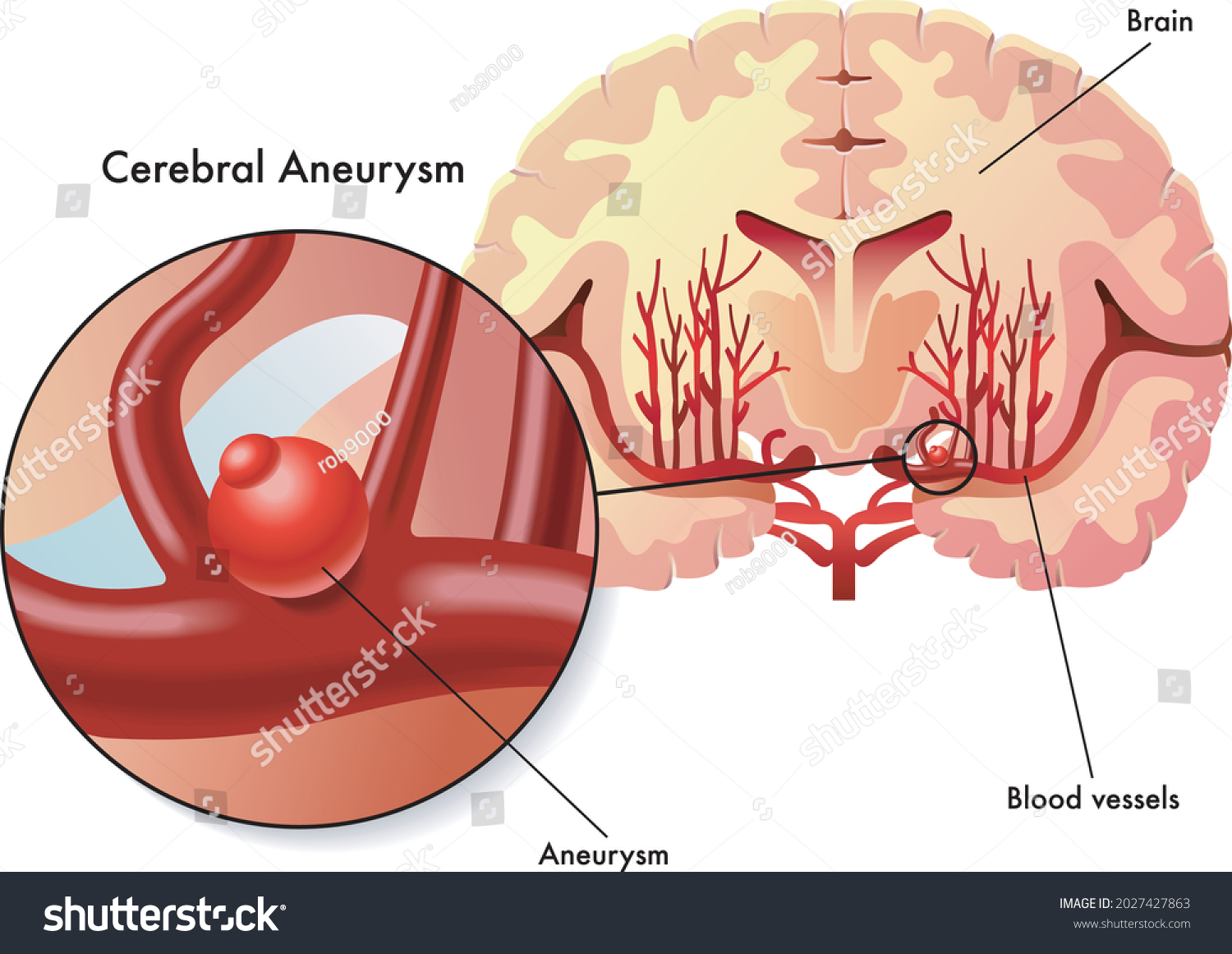
Der klinische Schwerpunkt „Vaskuläre Neurochirurgie“ befasst sich mit der Behandlung von Patient:innen mit Gefäßerkrankungen des Gehirns und Rückenmarks, wie z.B. Aneurysmen, Angiomen, Kavernomen, arteriovenösen Fisteln und intrazerebralen Blutungen.
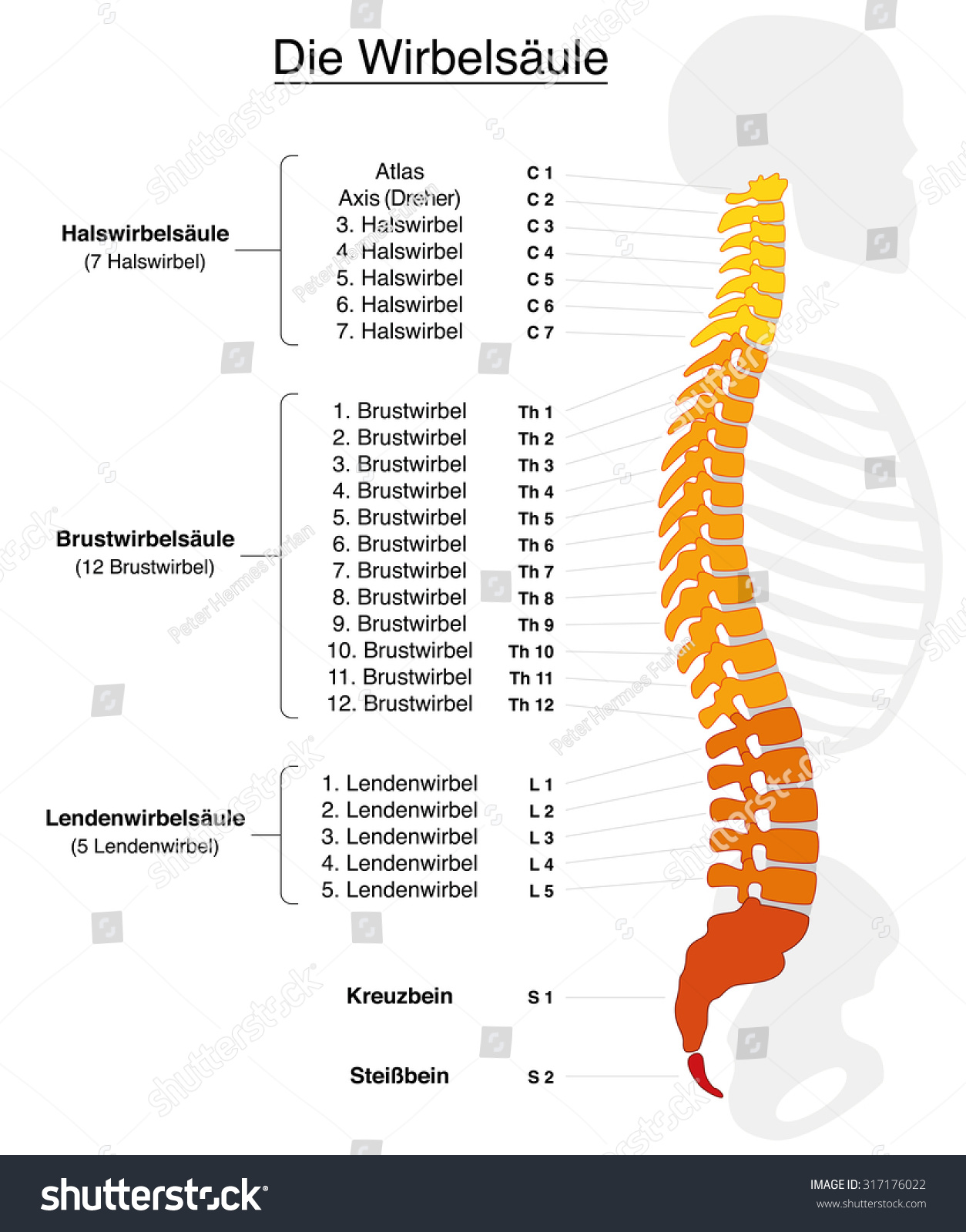
Die Wirbelsäule (lat. columna vertebralis) besteht aus 24 freien Wirbeln (lat. vertebra), die über 23 bewegliche Bandscheiben (Zwischenwirbelscheiben, lat. discus intervertebralis) verbunden sind, sowie 8 bis 10 Wirbeln, die zu Kreuz- und Steißbein verwachsen sind.
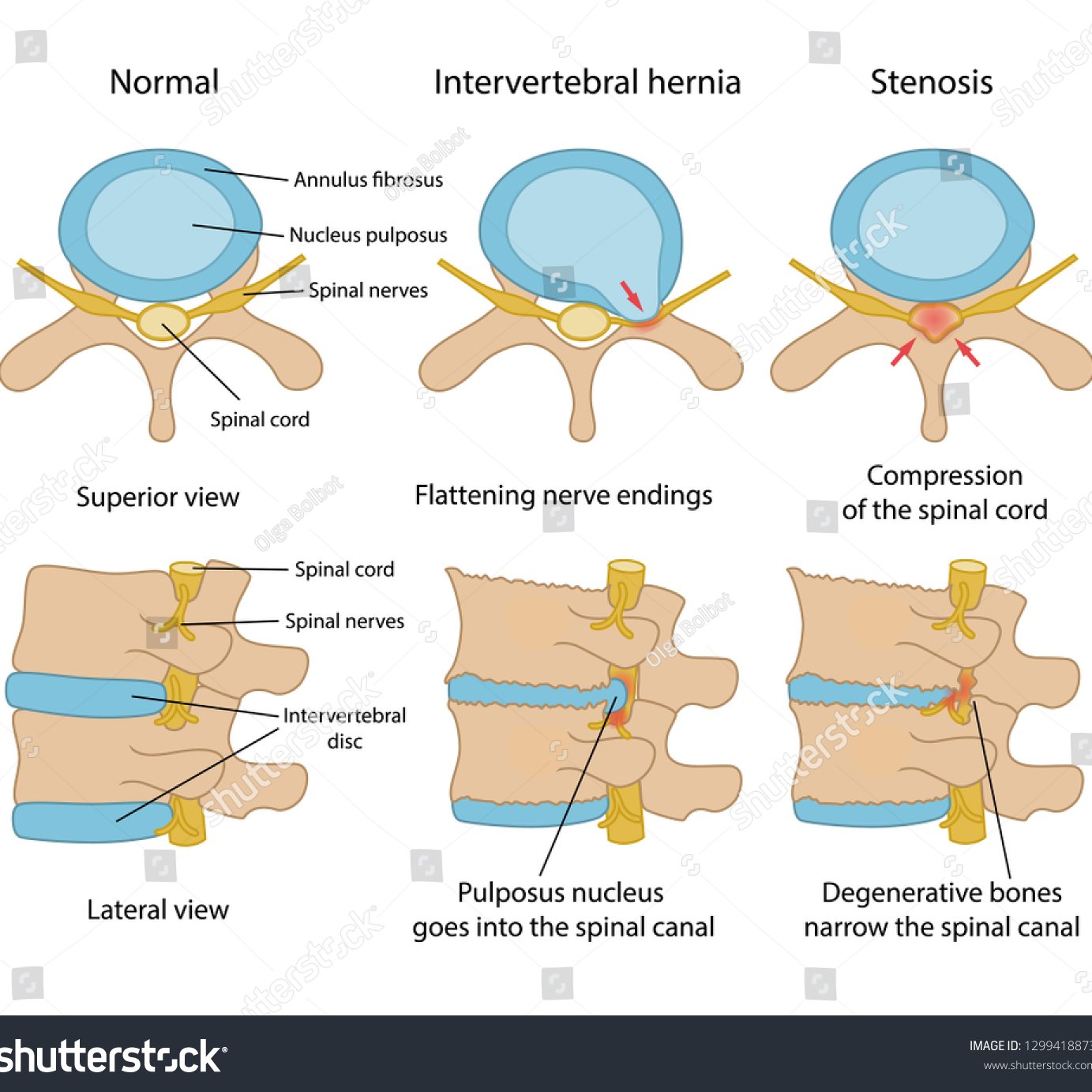
Degenerative Erkrankungen der Wirbelsäule umfassen lumbale (an der Lendenwirbelsäule-LWS), thorakale (an der Brustwirbelsäule-BWS) oder zervikale (an der Halswirbelsäule-HWS) Bandscheibenvorfälle und Spinalkanalstenosen, sowie Wirbelsäulentumore und Spondylolisthesen oder Wirbelgleiten – eine Überbeweglichkeit von Wirbelkörpern, die sich gegeneinander verschieben und ebenfalls eine Einengung (Stenose) des Wirbelkanals bewirken.

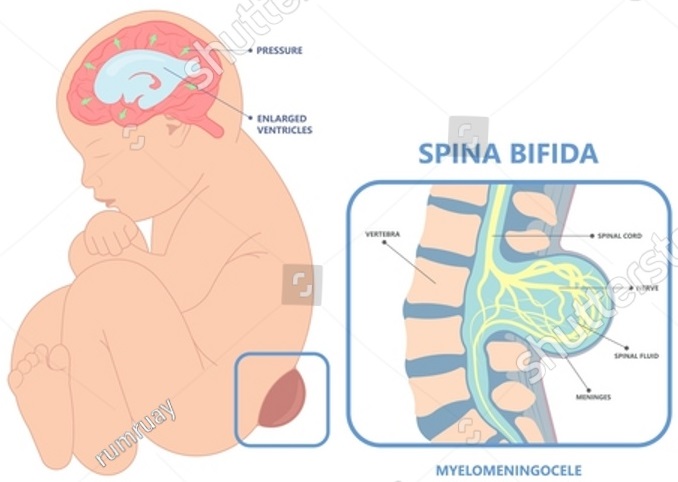

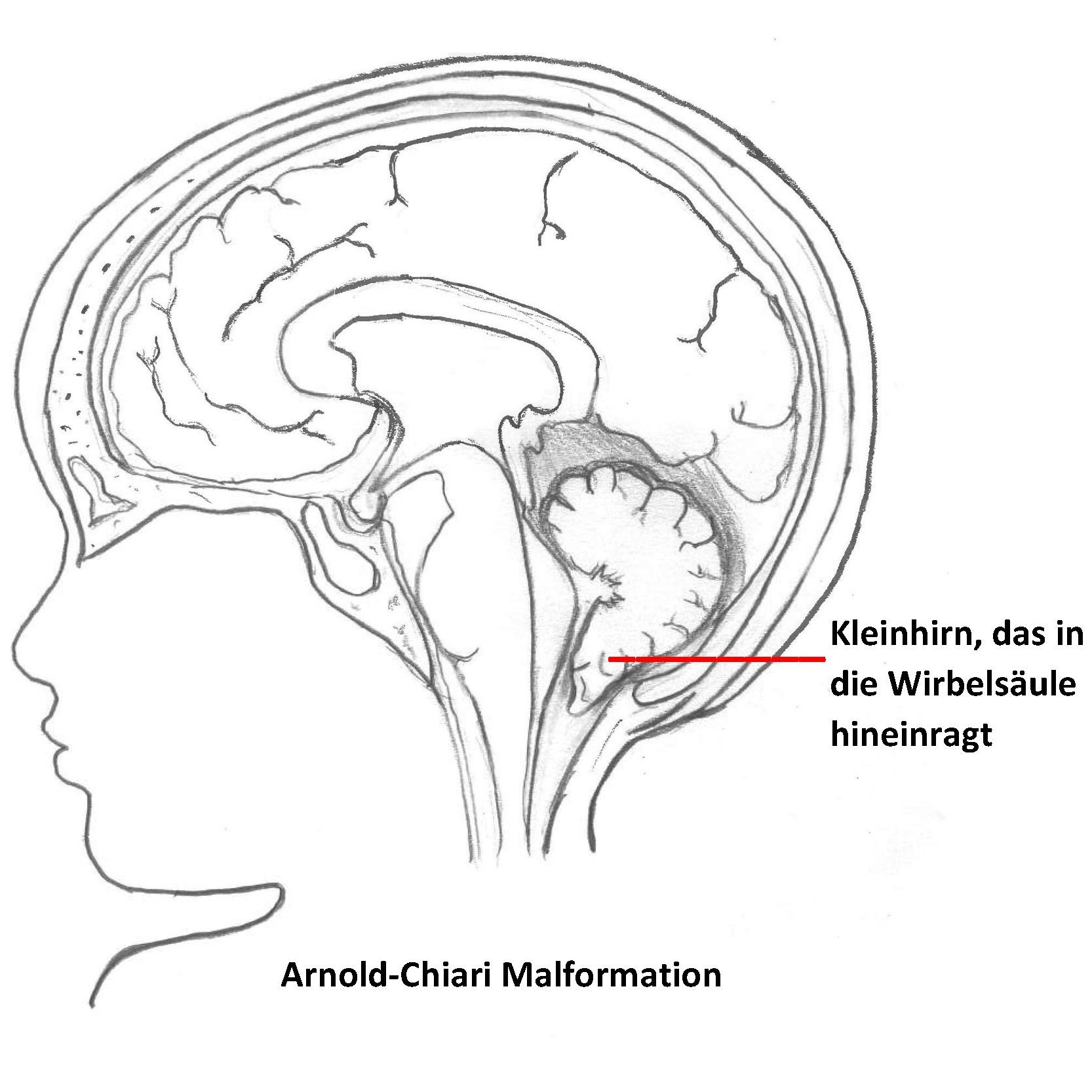
Das Leistungsspektrum unserer Klinik wird auch durch die neurochirurgische Behandlung funktioneller Störungen im Bereich des Nervensystems wie Neuralgien, Schmerzen (z.B. Cluster-Kopfschmerzen), Spastik und Bewegungsstörungen komplettiert. In weiterer Folge ist auch die Psycho-Neurochirurgie zu erwähnen, die Medikamententherapie-refraktäre psychisch-funktionelle Störungen wie Depressionen oder Zwangsstörungen mit reversiblen, nicht zerstörerischen Eingriffen am Gehirn behandelt.